
2024-03-03 04:20:02
Secuenciación del genoma completo y variantes genómicas.
Cuadro 1 Características de los 59 aislados de M. tuberculosis.
Linajes
Resistencia a las drogas
Treinta aislados (50,1%) fueron resistentes a cualquier medicamento contra la tuberculosis, incluida isoniazida (15/59, 25,4%), rifampicina (7/59, 11,8%), tuberculosis MDR (5/59, 8,4%) y pre -XDR (1/59, 1,7%) (Tabla 1). Se generaron datos fenotípicos de susceptibilidad a los fármacos para rifampicina e isoniazida, y se confirmaron los perfiles de resistencia genotípicos. Las frecuencias de las mutaciones DR se compararon con las de los conjuntos de datos públicos de Tailandia («otra Tailandia», n = 1456) y «Global50k» (n = 50,722) (Tabla 2; Tabla S1). Las mutaciones más frecuentes que subyacen a la resistencia a la isoniazida fueron katG Ser315Thr (9/59; otros Tailandia 56,9%; global 78,6%) y fabG1 -15C>T (5/59; otros Tailandia 6,1%; global 25,0%). Para la rifampicina, estaban presentes las mutaciones del codón rpoB 450 Ser450Trp y Ser450Leu (5/59; otros Tailandia 36,4%; global 58,8%). Se identificaron algunas mutaciones que se consideran menos prevalentes, concretamente para la etionamida (ethA Thr232Ala 5/59) y la pirazinamida (pncA Ser104Arg 5/59). Las mutaciones subyacentes para la resistencia a ciprofloxacina o fluoroquinolonas fueron gyrA Asp94Gly (1/59) y gyrB Asp494Ala (1/59). También se detectó la mutación thyX -16C>T ligada a PAS (1/59). También se encontraron varias mutaciones potenciales de estreptomicina (rpsL Lys43Arg, rpsL Lys88Arg, rrs 514A>C, rrs 517C>T) (Tabla 2).
Tabla 2 Mutaciones de resistencia a medicamentos identificadas en los 59 aislados de M. tuberculosis.
Utilizando los datos de la ONT, buscamos variantes estructurales en genes conocidos resistentes a los medicamentos (Tabla 3). Se identificaron eliminaciones en 10 aislamientos y se confirmaron con datos de Illumina. El tamaño de las eliminaciones identificadas fue similar en todas las plataformas (rango: ONT 35–10,981 pb; Illumina 27–10,983 pb), y las de Rv3083, Rv1258c, eis y thyA cubren casi todos sus genes. Un aislado (S27) tenía un gen eis eliminado. Se ha descubierto que la sobreexpresión de eis es una de las principales causas de resistencia al antibiótico kanamicina, y su eliminación completa podría mejorar la susceptibilidad de M. tuberculosis a los antibióticos, una inversión del mecanismo típico de DR9. Además, identificamos varios aislados con deleciones hacia el final del gen fbiC, que pueden influir en la eficacia de ciertos antibióticos10. Algunos estudios han indicado un vínculo entre las mutaciones que ocurren cerca del final del gen y un aumento en la CIM de la bacteria y una mayor resistencia al delamanid11. Estos hallazgos subrayan la necesidad de realizar más investigaciones sobre las implicaciones de estas eliminaciones del FBIC.
Tabla 3 Eliminaciones encontradas utilizando la plataforma Oxford Nanopore Technology.
Análisis filogenético y transmisión.
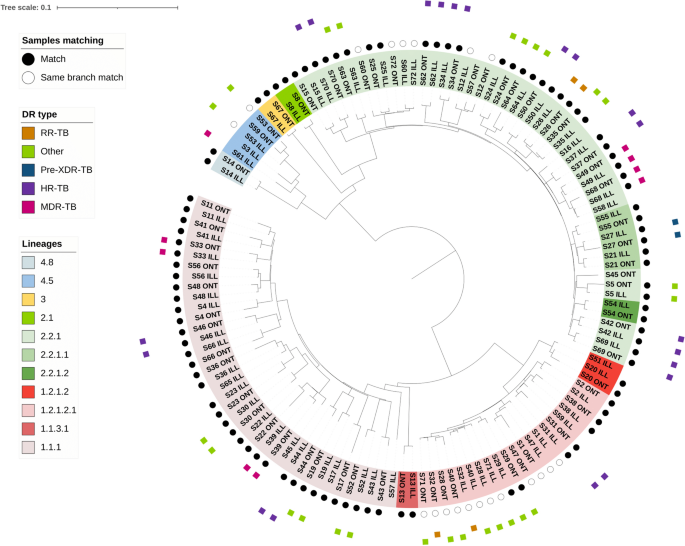
Utilizando los SNP detectados en todas las muestras, un análisis filogenético reveló la agrupación esperada por linaje y que los aislados replicados se emparejaron en el árbol, lo que indica una divergencia mínima entre los aislados secuenciados por ONT y Illumina. Sin embargo, los aislados secuenciados con ONT tienden a demostrar longitudes de rama marginalmente alargadas (Fig. 1). En un análisis combinado con los otros aislados de Tailandia (n = 1456), con un umbral de similitud de SNP de como máximo 20 SNP de diferencia, se identificaron 153 grupos, con tamaños que oscilaban entre 2 y 224, y un tamaño mediano de 2 (Fig. 2). ). Siete grupos incluyen 15 aislados de estudio, y cinco grupos contienen exclusivamente dos aislados de estudio cada uno. Otro grupo (n = 41) contenía dos aislamientos del estudio y estaba formado por al menos muestras de TB-MDR procedentes de la región de Roi Et. El otro grupo (n = 7) tenía 3 aislamientos de estudio con sensibilidad a los medicamentos (todos de Chiang Rai). En umbrales de diferencias de 5 y 10 SNP, hubo 134 (rango de tamaño: 2 a 150 muestras) y 154 (rango de tamaño: 2 a 213 muestras) grupos, respectivamente. En estos umbrales de SNP más estrictos hubo al menos 17 aislamientos de estudio dentro de grupos de tamaños 2 o más, incluidos dos de los aislamientos de nuestro estudio dentro del clado dominado por TB-MDR.
Figura 1
Análisis de árboles filogenéticos. El árbol filogenético revela un alto grado de concordancia y agrupación de réplicas secuenciadas utilizando las plataformas Oxford Nanopore Technologies (ONT) e Illumina.
Figura 2
El análisis combinado de Tailandia de los aislamientos de nuestro estudio (n = 59; cuadrados) y otros aislamientos de Tailandia (n = 1456; círculos) revela grupos de alta similitud. Aplicamos un valor de corte de 20 SNP o menos diferencias entre aislados vinculados. Los cuadrados son muestras incluidas en este estudio y la imagen se creó utilizando la herramienta “tgv” (https://github.com/jodyphelan/tgv).
Variantes estructurales en genes pe/ppe
Los genes Pe/ppe son regiones genéticas muy variables dentro del genoma de M. tuberculosis (~ 10%) que están implicadas en interacciones con el huésped humano12. Debido a su naturaleza altamente variable, normalmente se eliminan del análisis. Sin embargo, utilizando datos de secuenciación de ONT, identificamos 830 eliminaciones en 56 genes pe/ppe en todas las muestras (Tabla S4). El análisis reveló varias eliminaciones específicas de linaje. Por ejemplo, la eliminación completa del gen ppe50 en las cepas L1 (Fig. 3), la eliminación de ppe66/67 en las cepas L3 (Tabla S4) y pequeñas eliminaciones en pe_pgrs2 y pe_pgrs6 que eran específicas de L1.2.1.2.1. Había complejidad en el gen ppe8, con deleciones en el inicio del gen para las cepas L1 y en la región RD304 abarcada para L2. También hubo eventos de fusión genética, donde todas las cepas L2 tenían deleciones de los genes pe_pgrs3 y pe_pgrs4 en sus respectivos límites genéticos. Se encontró que varios genes pe/ppe, incluidos ppe34 y pe_pgrs10, tenían pequeñas deleciones en la mayoría de los aislados del estudio. Nuestras variantes estructurales específicas de linaje corroboraron estudios anteriores que utilizaron datos de lectura larga de PacBio12 (Tabla S4) y subrayan la confiabilidad y reproducibilidad de nuestro enfoque y nuestros datos.
figura 3
#Secuenciación #multiplataforma #del #genoma #completo #para #aplicaciones #clínicas #vigilancia #tuberculosis